Terapia Genica e Charcot-Marie-Tooth

I vari approcci per le diverse forme di CMT
Le terapie geniche non sono una sola “cura” valida per tutte le CMT. Ogni forma genetica ha un meccanismo diverso: in alcuni casi manca una proteina, in altri ce n’è troppa, in altri ancora una proteina alterata diventa tossica per il nervo. Per questo la ricerca sta sviluppando strategie differenti, mirate al gene e al tipo di cellula coinvolta.

1. Prima di entrare nelle singole forme: tre idee chiave
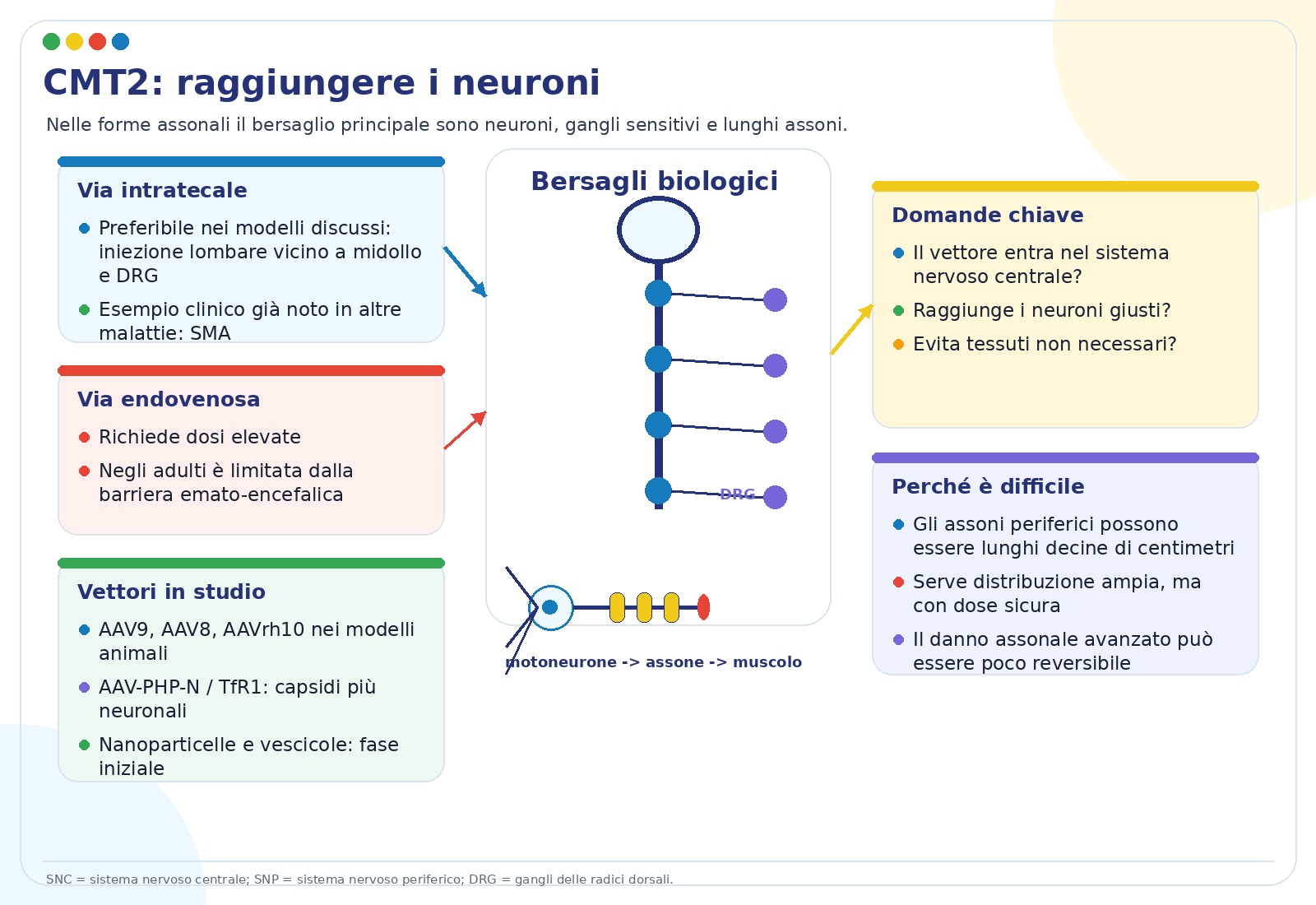
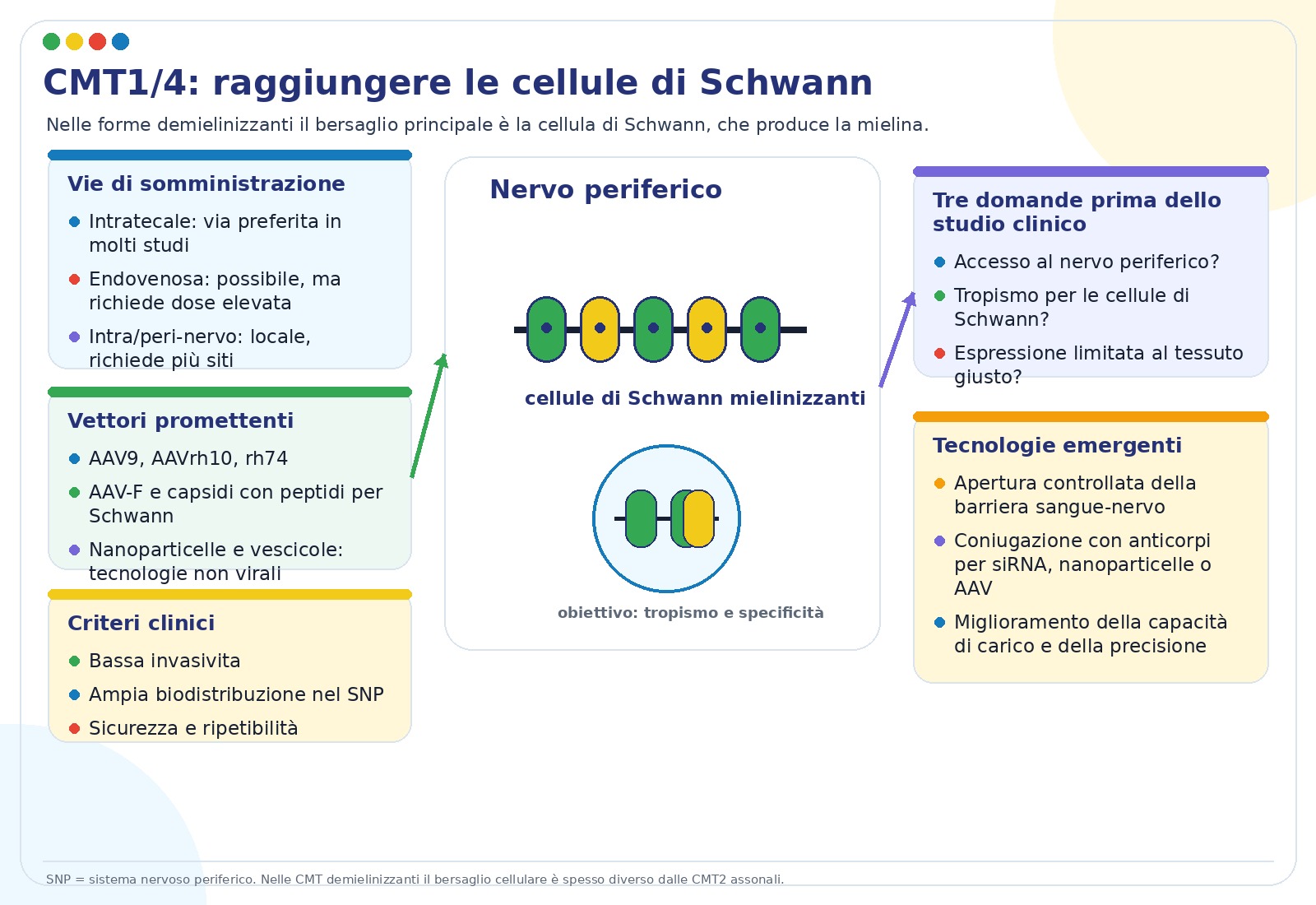
La CMT colpisce i nervi periferici, cioè i “cavi” che collegano midollo spinale, muscoli e pelle. Alcune forme danneggiano soprattutto l’assone, la parte lunga del neurone che trasporta il segnale; altre danneggiano soprattutto la mielina, il rivestimento isolante prodotto dalle cellule di Schwann. La terapia genica deve quindi raggiungere il bersaglio corretto.
Ridurre un gene troppo espresso
Nella CMT1A il problema principale è l’eccesso di PMP22. L’obiettivo è “abbassare il volume” del gene, senza spegnerlo troppo.
Sostituire un gene che non funziona
Nelle forme da perdita di funzione, come CMT1X, CMT4C, CMT4J o CMT2S, si cerca di portare una copia funzionante del gene nelle cellule giuste.
Neutralizzare un effetto tossico
In alcune forme dominanti, come CMT2A e CMT2D, una proteina alterata interferisce con la cellula. In questi casi si studiano silenziamento selettivo, “silence and replace” o strategie compensatorie.
Un punto è cruciale: la dose deve essere precisa. Troppo poco trattamento può non funzionare; troppo trattamento può creare nuovi problemi. Nella CMT1A, per esempio, ridurre PMP22 è utile solo entro un intervallo controllato, perché livelli troppo bassi di PMP22 sono associati a un’altra neuropatia ereditaria, la HNPP.
2. Dove devono arrivare le terapie
Le strategie per le CMT assonali, spesso indicate come CMT2, devono raggiungere soprattutto neuroni motori, neuroni sensitivi e assoni. Le strategie per le CMT demielinizzanti, come molte CMT1 e CMT4, devono invece raggiungere le cellule di Schwann, che formano la mielina nel sistema nervoso periferico.
3. Riassunto rapido per forma di CMT
La tabella sintetizza gli approcci discussi nel talk. La colonna “stato” non indica disponibilità clinica, ma il livello di sviluppo presentato nelle slide.
| Forma | Gene / meccanismo principale | Approcci in studio | Stato indicato nel talk |
|---|---|---|---|
| CMT1A | Duplicazione di PMP22: troppa proteina PMP22 nelle cellule di Schwann. | siRNA FALCON, siRNA con nanoparticelle di squalene, AAV-shRNA, AAV-miRNA, ASO, CRISPR/Cas9; possibile supporto con NT-3. | fase clinica FALCON/EDK060; altri approcci preclinici o NHP. |
| CMT1X | Mutazioni GJB1, proteina Cx32: perdita di comunicazione tra compartimenti della cellula di Schwann. | Sostituzione genica con AAV9, AAVrh10 o AAVrh74 guidati da promotori mielino-specifici; NT-3 come approccio trofico. | preclinico avanzato con dati in modelli murini e primati non umani. |
| CMT2A | Mutazioni dominanti di MFN2: alterazione di fusione, movimento e funzione dei mitocondri. | Silence and replace MFN2, aumento di MFN2 sano o MFN1, silenziamento allele-specifico, editing, modulazione di SARM1. | preclinico con risultati incoraggianti in cellule e modelli animali. |
| CMT2S / SMARD1 | Perdita di funzione di IGHMBP2: degenerazione dei motoneuroni, con forme gravi respiratorie nell’infanzia. | Sostituzione genica AAV9-IGHMBP2; ottimizzazione del promotore e della dose. | trial fase 1/2 indicato nelle slide come NCT05152823. |
| CMT2D | Mutazioni dominanti di GARS1: meccanismo tossico legato alla sintesi proteica e al metabolismo dei tRNA. | MicroRNA allele-specifici, terapia con tRNAGly, aumento di BDNF, possibili fattori trofici come NT-3. | preclinico / traslazionale. |
| CMT4J | Perdita di funzione di FIG4, coinvolto nella via endosoma-lisosoma. | Sostituzione genica AAV9-CBA.FIG4; programma ELP-02. | IND/NHP e primo studio umano pianificato nelle slide per metà 2026. |
| CMT4C | Perdita di funzione di SH3TC2: alterazione della maturazione delle cellule di Schwann e della mielina. | Sostituzione genica AAV9-hMPZmini.SH3TC2; possibile supporto con NT-3. | preclinico con studi dose-risposta e sicurezza in modelli murini. |
| CMT4A | Perdita di funzione di GDAP1. | Sostituzione genica AAV9-CBA.GDAP1. | fase iniziale secondo la panoramica del talk. |
| CMT4D | Perdita di funzione di NDRG1. | Sostituzione genica AAV9-MPZ.NDRG1, mirata alle cellule mielinizzanti. | fase iniziale. |
| CMT2E | Mutazioni di NEFL, con meccanismi variabili. | Editing CRISPR/Cas9 citato come direzione di ricerca. | fase iniziale. |
4. Gli approcci, forma per forma
CMT1A: ridurre PMP22 con precisione
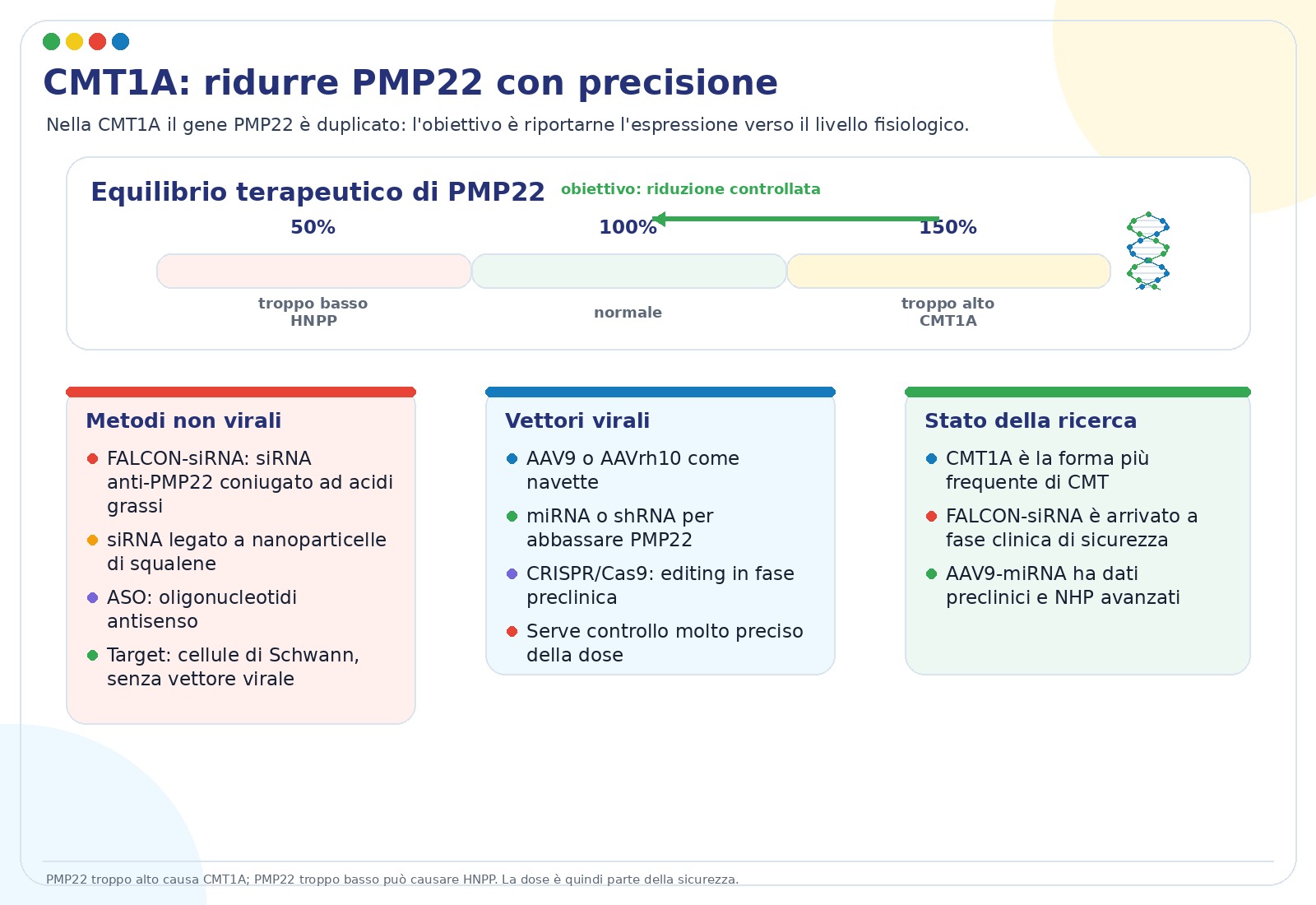
La CMT1A è la forma più frequente di CMT. Nelle slide viene indicata come circa 60% di tutte le CMT. È causata di solito da una duplicazione del gene PMP22: invece di produrre una quantità normale di proteina, la cellula ne produce troppa. Questa sovrapproduzione altera la mielina e rallenta la conduzione nervosa.
L’obiettivo terapeutico non è eliminare PMP22, ma riportarlo vicino a un livello fisiologico. È un equilibrio delicato: circa 150% di PMP22 è associato alla CMT1A, circa 100% è il livello atteso, mentre livelli troppo bassi possono portare a HNPP.
- siRNA FALCON anti-PMP22. Sono piccoli RNA progettati per ridurre la produzione di PMP22. La tecnologia FALCON li collega ad acidi grassi naturali per migliorare l’ingresso nelle cellule e la distribuzione nei tessuti. Nelle slide è riportato un programma di fase 1 con EDK060.
- siRNA con nanoparticelle di squalene. In modelli animali ha ridotto PMP22 e migliorato forza, mielinizzazione e parametri elettrofisiologici come CMAP e velocità di conduzione motoria.
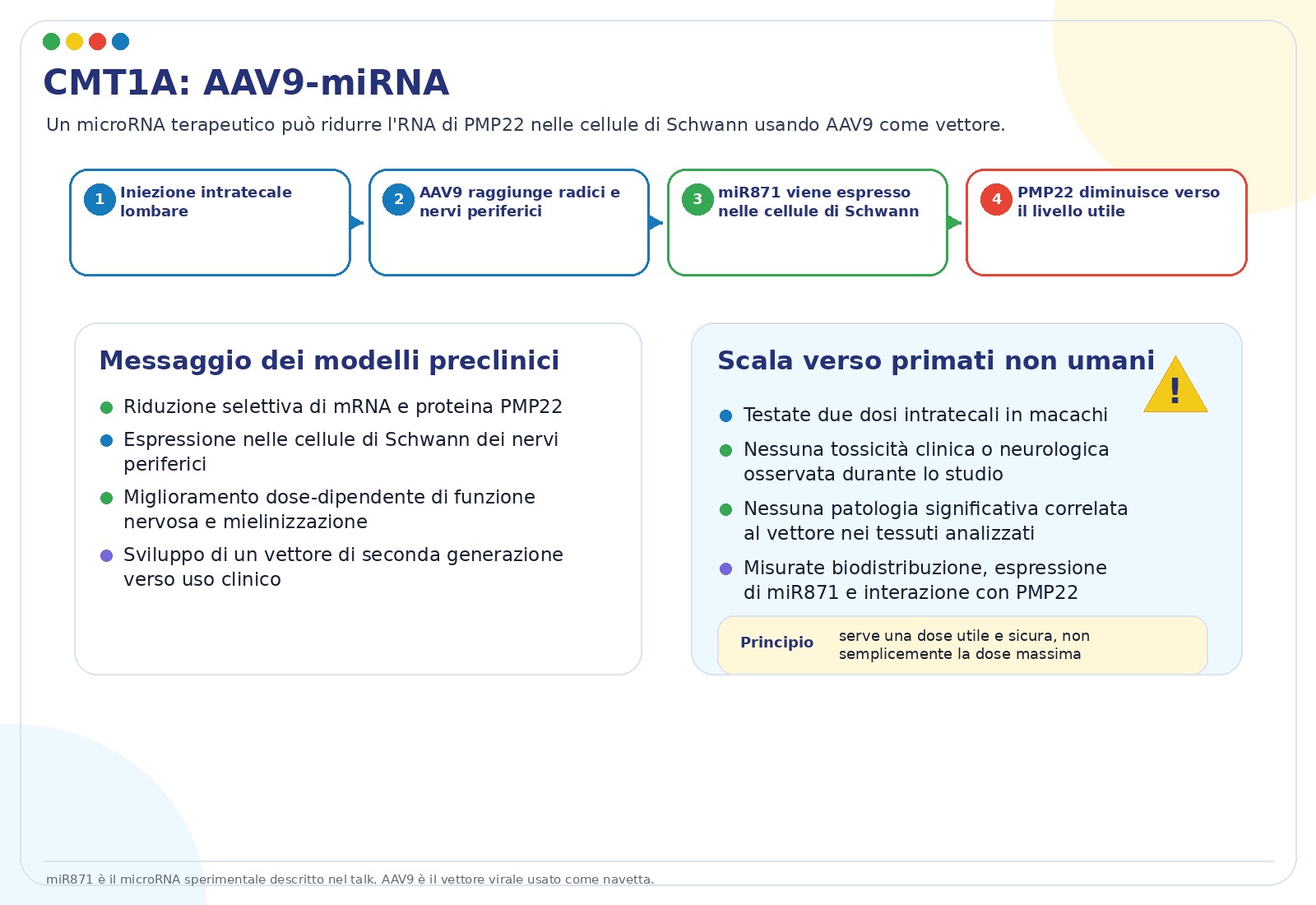
- AAV-shRNA e AAV-miRNA. Invece di somministrare direttamente l’RNA terapeutico, un vettore AAV porta nelle cellule l’istruzione per produrre molecole che abbassano PMP22. L’approccio AAV9-miR871 ha mostrato miglioramenti dose-dipendenti in modelli murini e dati di distribuzione/sicurezza in primati non umani.
- ASO e CRISPR/Cas9. Sono direzioni più sperimentali per modulare l’espressione o correggere il DNA, ancora lontane da un uso clinico ordinario.
CMT1X: sostituire GJB1/Cx32 nelle cellule di Schwann
La CMT1X è legata a mutazioni del gene GJB1, che produce connexina 32 o Cx32. Questa proteina forma canali di comunicazione nella cellula di Schwann. Quando Cx32 non funziona, la mielina e l’assone perdono stabilità nel tempo. La malattia è legata al cromosoma X: in genere i maschi sono colpiti prima e in modo più severo.
La strategia principale descritta nel talk è la sostituzione genica: un vettore AAV porta una copia funzionante di GJB1 nelle cellule di Schwann. Per limitare l’espressione alle cellule giuste, i ricercatori usano promotori mielino-specifici, come quelli derivati da MPZ.
- In modelli murini di CMT1X, AAV9-MPZ.GJB1 ha migliorato velocità di conduzione motoria, forza di presa, mielinizzazione, infiammazione e biomarcatori.
- Programmi con AAVrh10 e AAVrh74 hanno mostrato miglioramenti dose-dipendenti in modelli animali.
- Nei primati non umani, le slide riportano distribuzione nel sistema nervoso periferico ed espressione specifica nelle cellule di Schwann, senza segnali rilevanti di tossicità negli studi presentati.
CMT2A: proteggere gli assoni correggendo il problema dei mitocondri
La CMT2A è una delle forme assonali più frequenti. Le slide la indicano come circa 10% di tutte le CMT. È causata soprattutto da mutazioni dominanti di MFN2, un gene importante per la funzione dei mitocondri, le “centrali energetiche” della cellula.
Nei neuroni periferici, gli assoni possono essere molto lunghi. Per funzionare, hanno bisogno che i mitocondri si fondano, si muovano lungo l’assone e producano energia. Le mutazioni di MFN2 possono causare aggregazione dei mitocondri, riduzione del loro trasporto, stress cellulare, minore produzione di ATP e degenerazione assonale.
- Silence and replace. Una molecola shRNA riduce sia la copia mutata sia quella normale di MFN2; contemporaneamente il vettore introduce una versione di MFN2 modificata per resistere allo shRNA e funzionare correttamente.
- Aumento di MFN2 sano. In modelli murini, l’espressione neuronale di MFN2 normale tramite AAV9 intratecale ha migliorato contatti reticolo endoplasmatico-mitocondrio, morfologia mitocondriale, giunzioni neuromuscolari e funzione motoria.
- Aumento di MFN1. Poiché MFN1 e MFN2 lavorano insieme nella fusione mitocondriale, aumentare MFN1 può compensare in parte lo squilibrio causato da alcune mutazioni MFN2.
- Editing o silenziamento allele-specifico. Approcci mirati alla copia mutata sono concettualmente interessanti, ma complessi perché molte mutazioni sono diverse da famiglia a famiglia.
- Protezione dell’assone. Nella panoramica del talk compare anche SARM1, una via coinvolta nella degenerazione assonale, come possibile bersaglio neuroprotettivo.

CMT2S / SMARD1: sostituzione di IGHMBP2
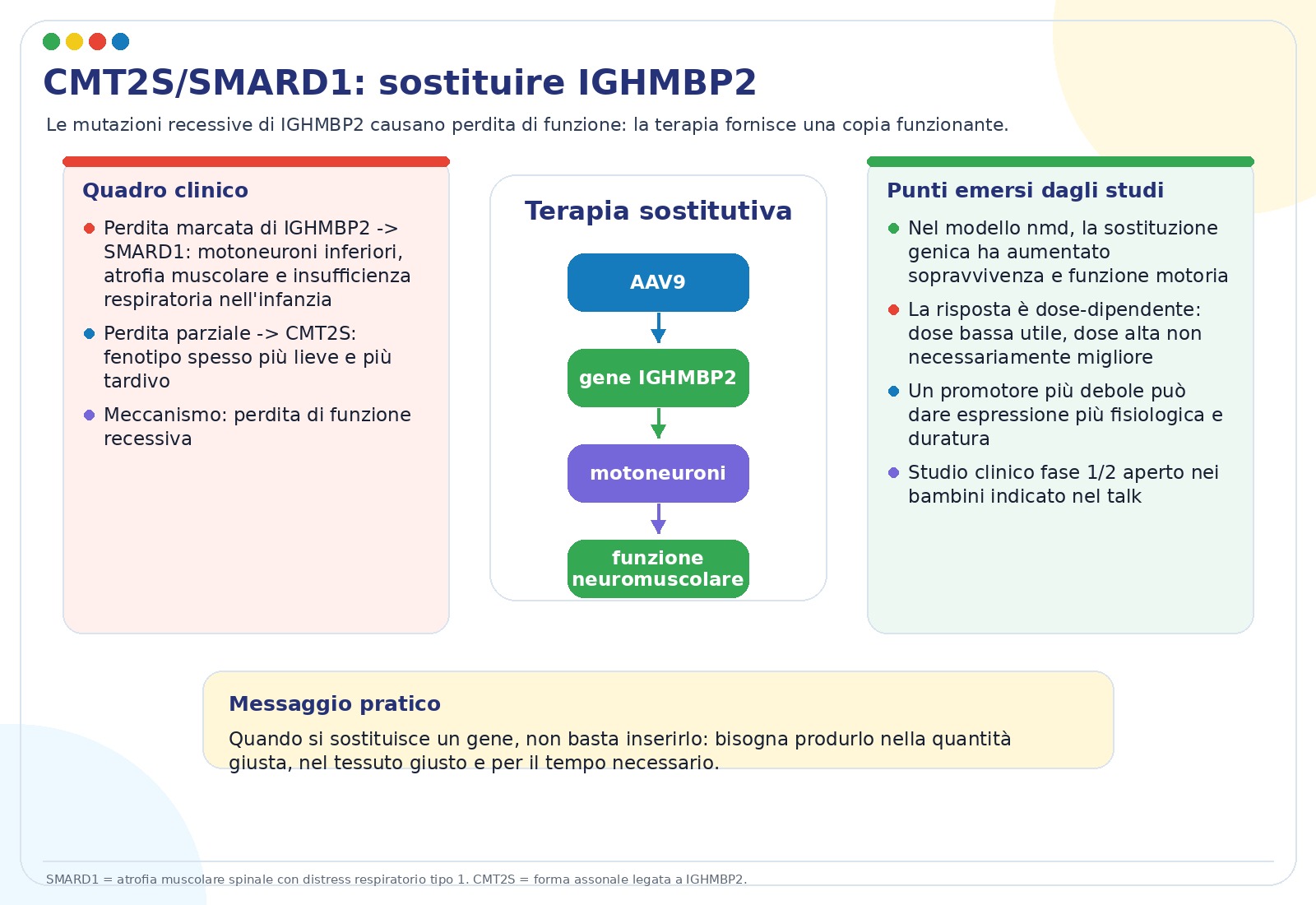
Le mutazioni recessive di IGHMBP2 possono causare un quadro molto grave chiamato SMARD1, con perdita dei motoneuroni, atrofia muscolare e complicanze respiratorie nell’infanzia. Quando la perdita di funzione è parziale, il fenotipo può essere più lieve e manifestarsi come CMT2S.
La logica terapeutica è diretta: se il gene non produce abbastanza proteina funzionante, si prova a fornire una copia sana tramite AAV9-IGHMBP2. Le slide sottolineano però un punto importante: la risposta è dose-dipendente e una dose troppo alta non è necessariamente migliore. In modelli murini, dosi più basse hanno migliorato sopravvivenza e funzione motoria più delle dosi elevate.
Secondo il talk, è in corso uno studio clinico di fase 1/2 open-label in bambini, indicato nelle slide come NCT05152823, con conclusione stimata nel 2030. È anche riportato che un promotore più debole, pensato per ottenere livelli più fisiologici di espressione, potrebbe offrire effetti a lungo termine migliori rispetto a promotori molto forti.
CMT2D: ridurre la tossicità di GARS1 o aumentare il tRNA corretto
La CMT2D è associata a mutazioni dominanti di GARS1, il gene per la glicil-tRNA sintetasi. Nelle slide è indicata come circa 0,4% di tutte le CMT. Il meccanismo non è una semplice mancanza di proteina: la proteina mutata acquisisce un effetto tossico, interferendo con il corretto uso dei tRNA e con la sintesi delle proteine.
Le strategie illustrate sono diverse:
- Silenziamento allele-specifico. MicroRNA artificiali sono progettati per spegnere la copia mutata di GARS1 lasciando attiva quella normale. Il vantaggio è la precisione; il limite è che non tutte le mutazioni sono facili da distinguere, soprattutto quando differiscono da quella normale per una sola base.
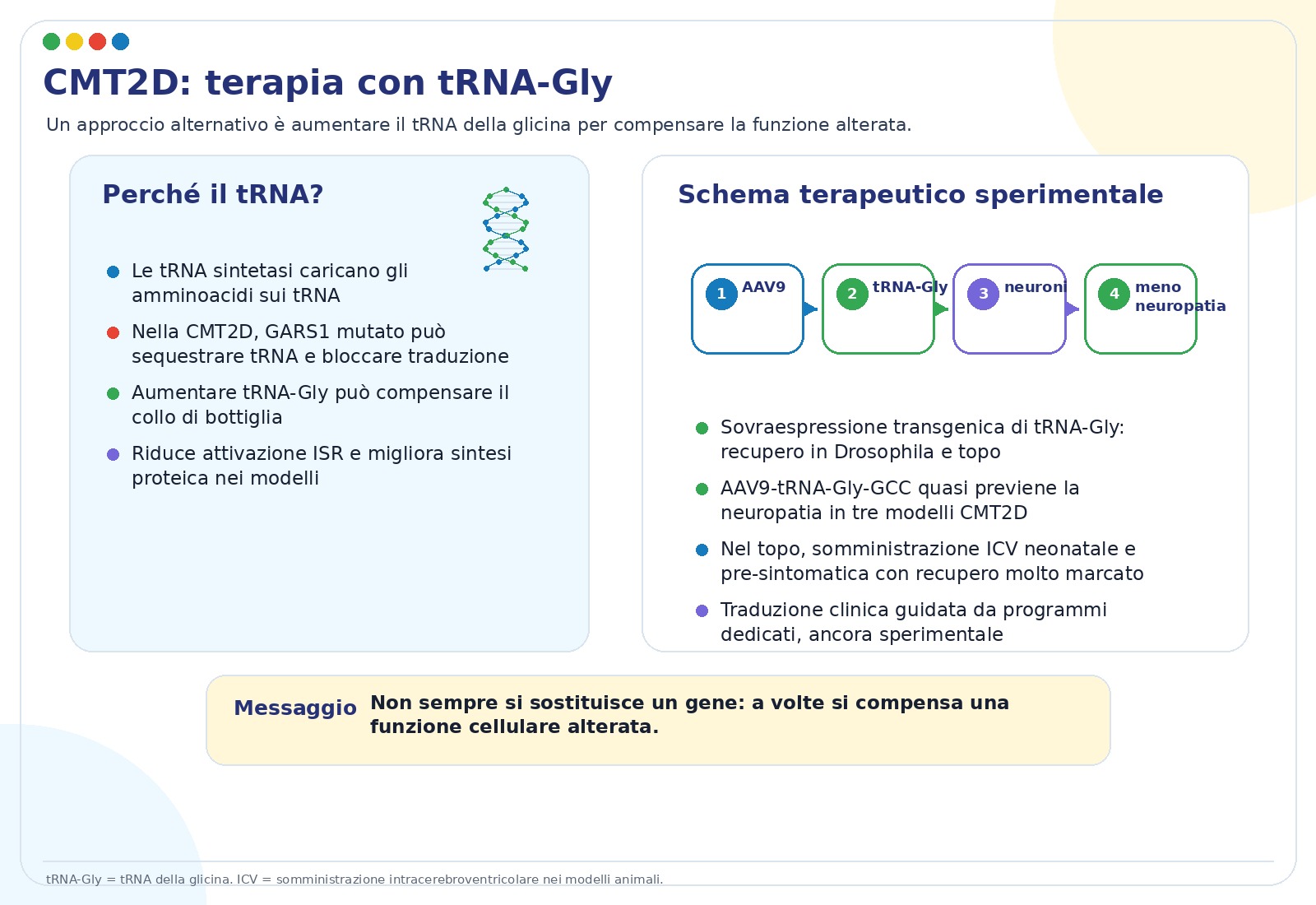
- Terapia con tRNAGly. Aumentare la disponibilità del tRNA della glicina può compensare il blocco causato dalla proteina mutata. In modelli animali, AAV9-tRNAGly ha prevenuto quasi completamente la neuropatia.
- BDNF. Alcune mutazioni interferiscono con il segnale BDNF/TrkB, importante per i neuroni. Aumentare BDNF nel muscolo o somministrarlo come proteina ha migliorato il trasporto assonale in modelli sperimentali.
- NT-3. Fattori trofici come NT-3 sono considerati approcci di supporto non specifici per più forme di CMT.
CMT4J: sostituzione del gene FIG4
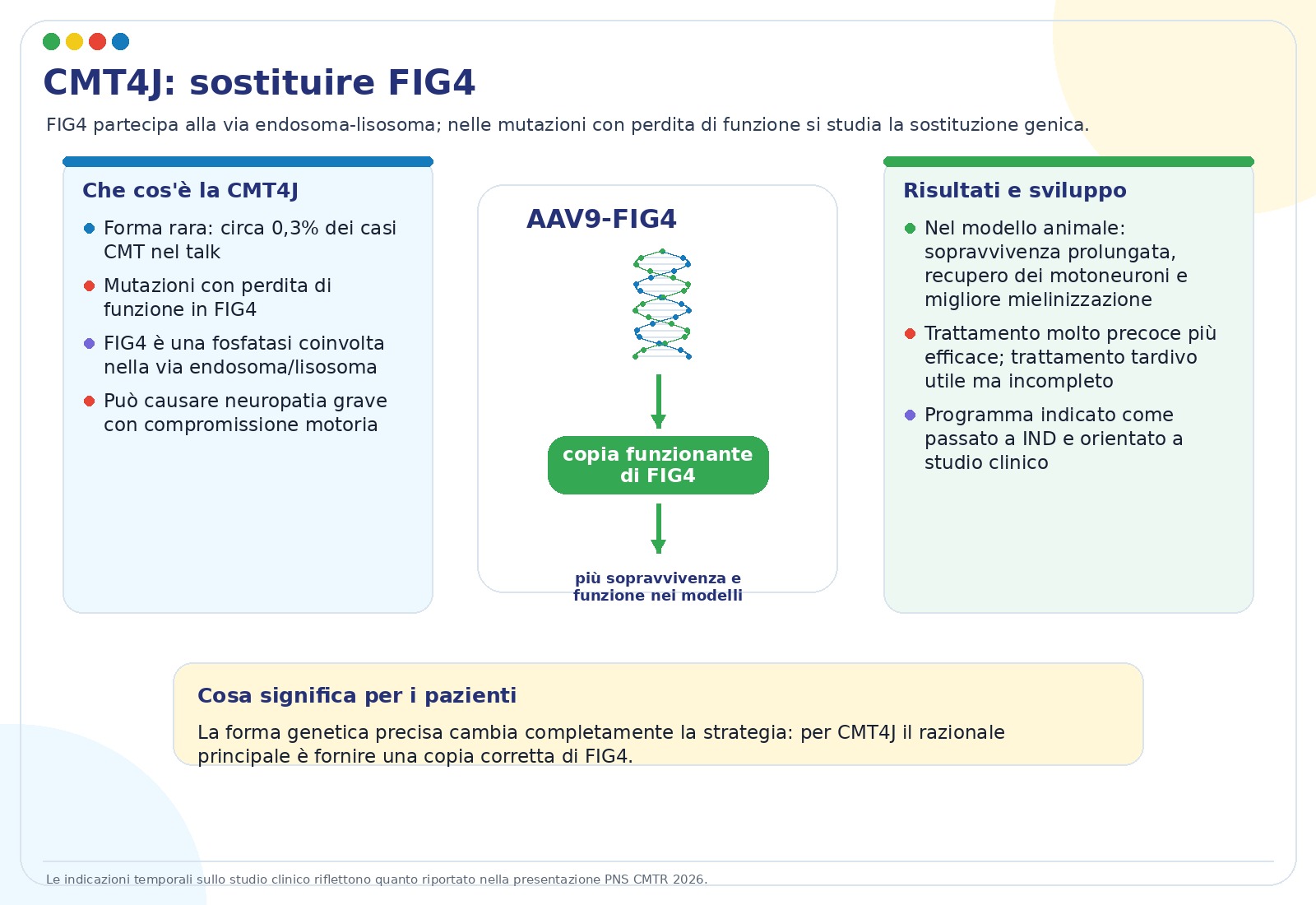
La CMT4J è una forma rara, indicata nelle slide come circa 0,3% di tutti i casi di CMT. È causata da mutazioni con perdita di funzione nel gene FIG4, importante per il sistema endosoma-lisosoma, cioè una rete cellulare che gestisce traffico, riciclo e degradazione di componenti cellulari.
La strategia più avanzata discussa è la sostituzione genica con AAV9-CBA.FIG4. Nei modelli murini, il trattamento precoce ha prolungato la sopravvivenza, migliorato la funzione motoria, favorito la protezione dei motoneuroni e migliorato la mielinizzazione. Il trattamento più tardivo ha dato benefici, ma incompleti: questo conferma l’importanza del tempo di intervento.
Le slide riportano che la terapia ha superato la fase IND e che il primo studio umano di fase 1/2 con ELP-02 era previsto per metà 2026, nel contesto di uno studio di storia naturale.
CMT4C: sostituire SH3TC2 nelle cellule di Schwann
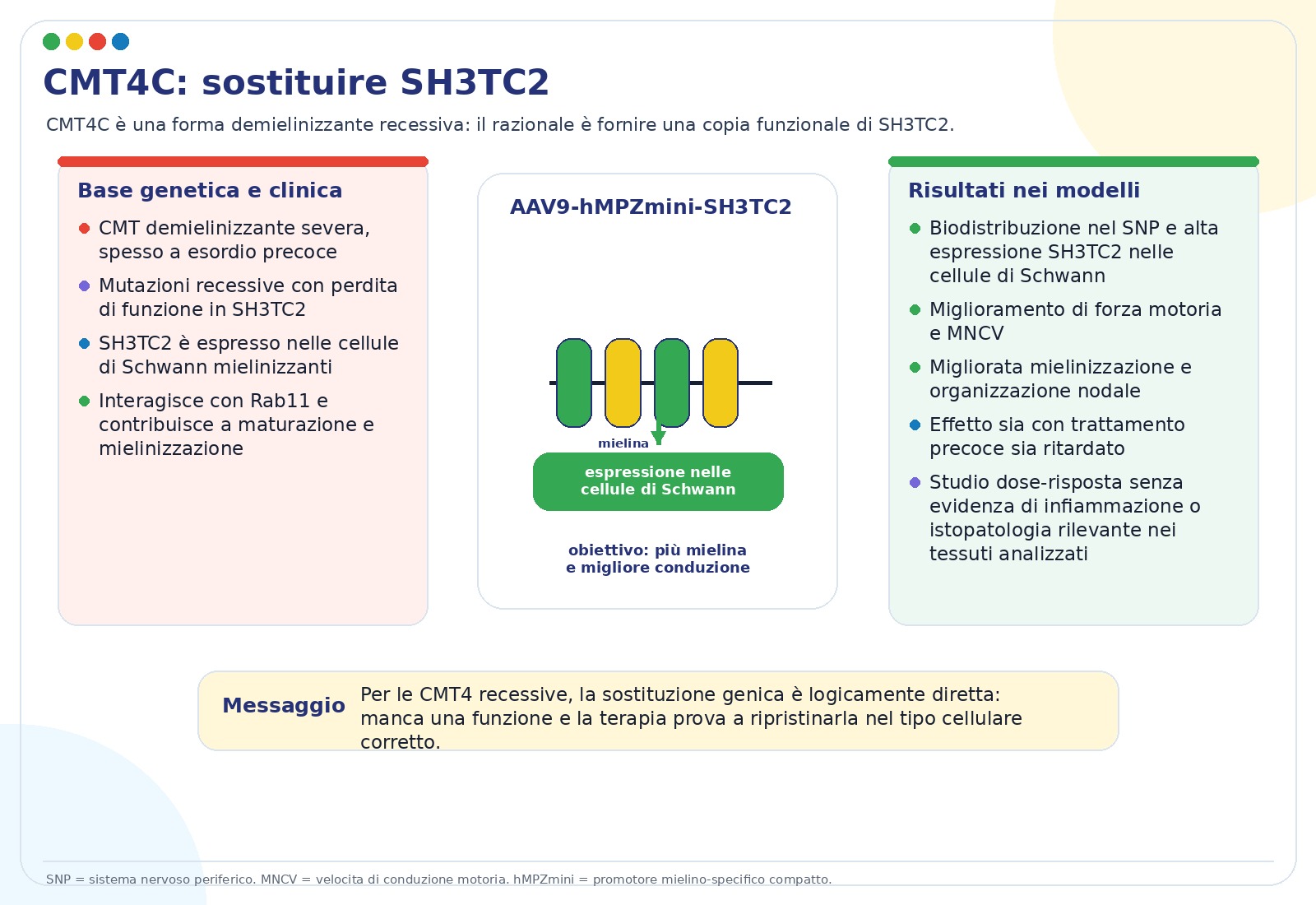
La CMT4C è una neuropatia demielinizzante spesso severa e a esordio precoce. Le slide la indicano come la forma CMT4 più frequente, circa 3–4% delle CMT demielinizzanti non-CMT1A. È causata da mutazioni recessive con perdita di funzione di SH3TC2.
SH3TC2 è espressa soprattutto nelle cellule di Schwann mielinizzanti e partecipa al traffico intracellulare legato a Rab11. Quando non funziona, la maturazione della cellula di Schwann e la mielinizzazione risultano alterate.
La strategia presentata è la sostituzione genica con AAV9-hMPZmini.SH3TC2, somministrata per via intratecale lombare in modelli murini. I risultati riportati includono migliore distribuzione del vettore nel sistema nervoso periferico, espressione nelle cellule di Schwann, miglioramento di forza muscolare, velocità di conduzione motoria, mielinizzazione e rimodellamento dei nodi di Ranvier. Le slide indicano efficacia sia con trattamento precoce sia con trattamento ritardato, con studi dose-risposta e assenza di alterazioni istopatologiche rilevanti nei tessuti analizzati.
Altre forme citate: programmi più iniziali ma razionali
Il talk cita anche forme per cui la logica terapeutica è chiara, ma i dati presentati sono più preliminari rispetto a CMT1A, CMT1X, CMT2A, CMT2D, CMT2S, CMT4C e CMT4J.
- CMT4A – GDAP1. Poiché molte mutazioni causano perdita di funzione, la direzione è la sostituzione genica con AAV9-CBA.GDAP1.
- CMT4D – NDRG1. La strategia citata è AAV9-MPZ.NDRG1, con promotore orientato alle cellule mielinizzanti.
- CMT2E – NEFL. Viene citato l’editing CRISPR/Cas9 come possibile approccio, da considerare ancora in fase iniziale.
- Neuropatie da aminoacil-tRNA sintetasi. Oltre a GARS1/CMT2D, sono citati YARS1, AARS1, HARS1, WARS1 e MARS1. Il meccanismo comune riguarda spesso un effetto tossico sulla sintesi proteica, sullo stress cellulare e sul citoscheletro dell’assone. Per alcune di queste forme, approcci trofici come BDNF sono stati studiati in modelli sperimentali.
- Approcci non specifici. NT-3 e BDNF non correggono direttamente il gene responsabile, ma puntano a sostenere la sopravvivenza, la funzione o la riparazione del nervo. Possono quindi essere studiati in più forme di CMT.
5. Perché la terapia genica nella CMT è difficile
Il messaggio finale del talk è prudente: la ricerca è avanzata, ma restano problemi tecnici e clinici importanti. Il nervo periferico è distribuito in tutto il corpo; alcune cellule bersaglio sono difficili da raggiungere; molti pazienti hanno una malattia lentamente progressiva, quindi i trial devono misurare cambiamenti piccoli ma significativi nel tempo.
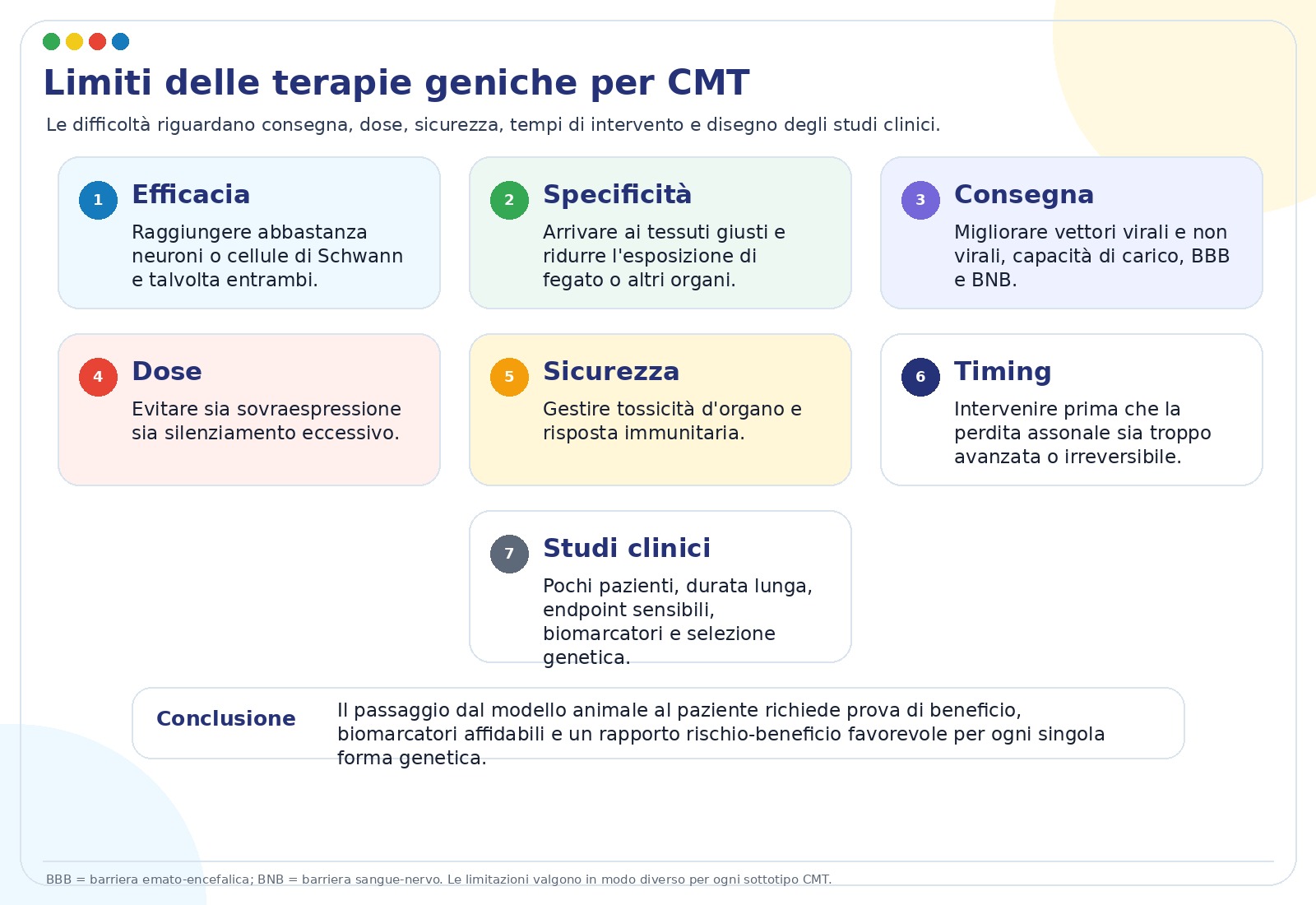
Problemi biologici e tecnici
- Raggiungere abbastanza neuroni o cellule di Schwann.
- Portare il trattamento nel tessuto giusto, evitando organi non bersaglio.
- Scegliere la via di somministrazione più efficace e meno invasiva.
- Controllare dose, durata dell’effetto e rischio immunitario.
Problemi clinici
- Intervenire prima che la perdita assonale sia troppo avanzata.
- Selezionare pazienti con diagnosi genetica precisa.
- Usare biomarcatori e scale cliniche sensibili.
- Organizzare trial sostenibili per malattie rare con pochi pazienti.