La Malattia di Charcot-Marie-Tooth (CMT)
AllegatiLa malattia di Charcot-Marie-Tooth (CMT) è una rara neuropatia ereditaria che colpisce i nervi periferici, causando debolezza muscolare e difficoltà motorie. Può manifestarsi a qualsiasi età, ha un andamento cronico e progressivo, ma spesso viene sottovalutata perché poco conosciuta. Informarsi sulla CMT è il primo passo per riconoscerla e migliorare la qualità di vita di chi ci convive
La malattia di Charcot-Marie-Tooth (CMT, spiegata in breve a questo link) è una patologia neurologica genetica ereditaria, classificata tra le malattie rare, che interessa i nervi periferici del controllo, del movimento e sensoriali. Fa parte delle malattie neuromuscolari, poiché danneggia indirettamente anche i muscoli sebbene sia una neuropatia periferica.
Cos’è la CMT?
È il disordine neurologico ereditario del sistema nervoso periferico più diffuso (l’incidenza è di 1 persona ogni 2.500), ma pochi la conoscono e la sanno riconoscere.

Charcot, Marie e Tooth furono i tre neurologi dell’800 da cui la malattia prende il nome, ma si può trovare anche sotto altri acronimi, quali ad esempio CM (Charcot-Marie), HMSN (Hereditary Motor and Sensory Neuropathie) o PMA (Peroneal o Progressive Muscolar Atrophy, o Atrofia Muscolare Peroneale).
È definita distale, perché colpisce le estremità dell’organismo, cioè gli arti inferiori (in particolare dal ginocchio al piede) e superiori (in particolare dal gomito alla mano). I primi ad ammalarsi sono i nervi e poi, per induzione, i sintomi coinvolgono tendini, muscoli e ossa. Solo in alcuni tipi di CMT vi è la compromissione del sistema nervoso centrale.
Può presentarsi a qualsiasi età, l’evoluzione è cronica ed è lentamente progressiva. Può manifestarsi diversamente per sintomi e gravità anche all’interno della stessa famiglia.
Cose importanti da sapere sulla CMT

10 cose che dovresti sapere sulla CMT
Gli esordi della malattia di Charcot-Marie-Tooth o CMT
Le manifestazioni della CMT sono molto variabili, anche all’interno della stessa famiglia. La progressione è per lo più lenta e spesso i primi sintomi vengono riferiti a cause non neurologiche, anche se riguardano in prevalenza l’indebolimento dei muscoli, che consegue alla degenerazione delle fibre nervose motorie. Solo in pochi casi la malattia si manifesta molto rapidamente, indebolendo la muscolatura dei piedi e delle gambe, per poi rimanere stazionaria per qualche decennio.
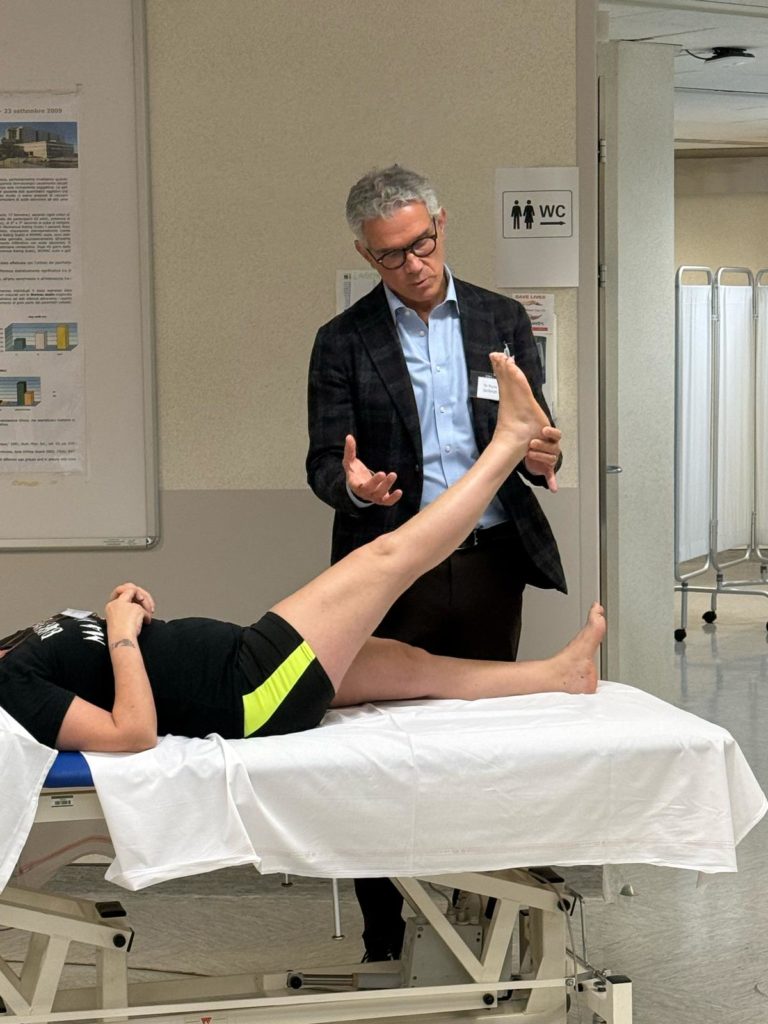
I sintomi della CMT
Sintomi ai Piedi e Gambe della CMT
Negli Arti Inferiori i sintomi più comuni sono: inciampo sull’avampiede, spesso per piccoli dislivelli e gradini (più frequente a piedi nudi); distorsioni di caviglia; distorsioni del ginocchio; goffaggine nel camminare; crampi ai polpacci. Spesso il paziente adatta spontaneamente il cammino, utilizzando scarpe con il tacco e facendo rialzare dal calzolaio la parte laterale esterna della suola, perché si consuma di più. Con il passare del tempo, l’indebolimento della flessione dorsale dei piedi si accentua e il paziente è costretto a sollevare le ginocchia più del normale, per evitare di inciampare con la punta dei piedi: questo cammino – che ricorda quello del cavallo – è detto deambulazione “steppante” o equina, ed è piuttosto stancante.
Raramente la malattia si diffonde ai muscoli delle cosce; quando questo avviene la conseguenza è lo scarso controllo del ginocchio e conseguenti cadute, che possono portare alla decisione di utilizzare la sedia a rotelle.
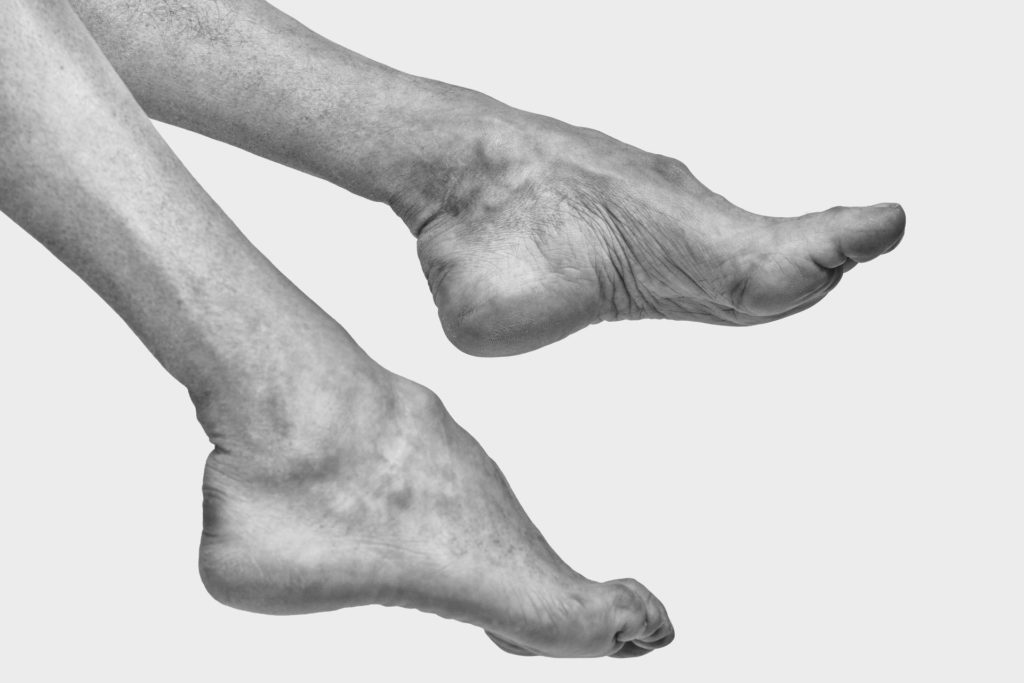
Sintomi alle Mani e Braccia della CMT
Per quanto riguarda gli Arti Superiori, l’inizio dei sintomi è più tardivo e spesso l’indebolimento è talmente lieve, che non determina un deficit funzionale. I disturbi più frequentemente lamentati sono difficoltà ad abbottonarsi e a sbottonarsi, a usare chiusure lampo, a cucire, a scrivere calcando, a girare la chiave, a svitare tappi e coperchi di barattoli. Questi problemi si accentuano con il freddo, che comporta anche una sensazione molesta alle gambe, un peggioramento dell’equilibrio e della sensibilità fine. L’alcool e alcuni farmaci possono causare peggioramento.

Il dolore neuropatico non è frequente (a parte quello dei crampi). Quello percepito, nella maggior parte dei casi, non è dovuto alla neuropatia in se, ma alle conseguenze che provocano la patologia e l’instabilità motoria sull’apparato osteoarticolare (poco equilibrio, deformità dei piedi e delle ginocchia, artrosi, esiti di traumatismi). L’indebolimento dei muscoli si accompagna al loro assottigliamento (atrofia muscolare).
Sebbene dopo i 50 anni di età si verifichi un lento peggioramento, nella maggior parte dei casi la disabilità non è grave. La malattia non riduce la durata della vita, ma ne peggiora sensibilmente la qualità.
Una deformità tipica, ma non esclusiva della CMT, è il piede cavo, presente nella maggioranza dei casi. Nel 10% dei casi vi è cifoscoliosi. Esistono infine forme rarissime, in cui si indeboliscono anche i muscoli respiratori e quelli della fonazione, con paralisi delle corde vocali, e anche forme in cui sono compromessi altri organi e apparati (specie l’orecchio, con sordità neurosensoriale, e l’occhio, con atrofia ottica).
Certamente la ricerca dovrà percorrere ancora molta strada per avere un quadro completo e certo delle caratteristiche sintomatologiche della CMT, ma in ogni caso si può dire che solo in poche situazioni, si presenta una disabilità grave, tanto da costringere all’uso della sedia a rotelle. ACMT-Rete è costantemente impegnata nel migliorare la qualità di vita delle persone con CMT.
Diagnosi della malattia di Charcot-Marie-Tooth o CMT
La diagnosi di sospetta malattia di Charcot-Marie-Tooth parte dall’osservazione clinica e si avvale principalmente di un esame elettrofisiologico ai quattro arti. È ormai fondamentale il test genetico, che si fa con un semplice prelievo di sangue, per la ricerca di mutazioni fra i numerosi geni coinvolti: ciò consente la diagnosi precisa in oltre il 60% dei casi. Tuttavia, la lista dei geni è destinata ad allungarsi, se ne conoscono già più di 90 e nel tempo le metodiche di indagine di analisi del DNA di nuova generazione permetteranno un’analisi sempre più completa. La biopsia del nervo dovrebbe essere sempre l’ultima indagine, da considerarsi solo in casi particolari e quando gli esami precedenti non abbiano identificato alcuna delle forme di CMT rilevabili con i test genetici a disposizione.

Molti casi gravi si notano nei figli di persone che, al momento del concepimento, non avevano ancora manifestato alcun sintomo oppure i cui genitori erano i cosiddetti “portatori sani” (ovvero forme recessive di CMT). L’identificazione genetica dei vari tipi di CMT permette al paziente di pianificare il proprio futuro, decidendo per un’eventuale procreazione assistita, e comporta anche una maggiore consapevolezza per la gestione del trattamento riabilitativo.
Le possibilità di trattamento attualmente disponibili per la malattia di Charcot-Marie-Tooth o CMT
Attualmente non esiste una terapia farmacologica. Per la forma più comune (CMT1A) numerosi trial clinici hanno studiato una terapia con acido ascorbico (vitamina C), ma purtroppo sono stati completati con esito negativo. Vi sono, tuttavia, numerosi sforzi per disegnare sperimentazioni più efficaci e la ricerca continua su una serie di farmaci tra cui lonaprisan o la niacina (che sembra adatta alle CMT ipermielinizzanti) e nuovi farmaci inibitori dell’istone deacetilasi hanno dimostrato un’efficacia nei modelli animali con CMT.
Al momento i pazienti possono convivere meglio con la CMT soltanto grazie alla chirurgia e alla riabilitazione, che finora ha dato ottimi risultati. Anche in questo campo, le conoscenze sono ancora insufficienti e poco diffuse a individuare percorsi mirati e utili a impedire la progressione della neuropatia.
Le persone con CMT non devono sottovalutare i problemi legati all’equilibrio ed evitare di cadere, perché lunghi periodi di immobilizzazione possono essere deleteri per il sistema muscolare già compromesso. L’utilizzo di ortesi e ausili come scarpe adatte, opportunamente modificate, di plantari ben confezionati ed eventualmente di tutori di caviglia, migliora notevolmente l’equilibrio e il cammino. Una moderata attività fisica può essere utile, un’attenta chirurgia del piede può migliorare l’equilibrio e il cammino.
Il dott. Pareyson, uno dei neurologi di riferimento in Italia, ha tenuto un webinar di approfondimento sulla CMT, lo trovate a seguire 👇
Le testimonianze dei pazienti con CMT
Sul nostro canale YouTube trovi numerose testimonianze e racconti di persone con CMT, come quella di Elisa. Non sei solo/a, contattaci e ti aiuteremo a gestire al meglio la convivenza con questa malattia❣️
Per ulteriori approfondimenti sulla malattia di Charcot-Marie-Tooth o CMT e sulle varie forme esistenti, consultare gli allegati:
I Lipidi come nuovi biomarcatori e potenziale bersaglio per la CMT

Abbiamo chiesto al Dott. Alessio Silva, neo-dottore di ricerca presso l’Università di Leuven, di raccontarci i risultati delle sue ricerche sul ruolo dei lipidi, la componente “grassa” che soprattutto nelle cellule nervose, che ne sono ricche, potrebbero portare alla scoperta di nuovi biomarcatori e trattamenti della CMT Piacere, Alessio Mi chiamo Alessio Silva, sono originario […]
La Ricerca Clinica, le Sperimentazioni e le Sfide per la CMT

Lo sviluppo di nuove terapie rappresenta un processo scientifico estremamente rigoroso, lungo e costoso, che richiede una media di 12 anni e investimenti di milioni di euro per passare dalla scoperta molecolare all’approvazione di un farmaco. Questo iter complesso è stato analizzato nel dettaglio durante un recente seminario educativo promosso dalla European CMT Federation (ECMTF), […]
Il Ruolo dell’Attività Fisica Adattata nella Malattia di Charcot-Marie-Tooth

In un recente lavoro intitolato “La malattia di Charcot-Marie-Tooth: ruolo dell’attività fisica adattata e del ricondizionamento muscolare nella gestione funzionale”, pubblicato sulla Rivista di Neurologia e che qui riassumiamo, gli autori Michele Perniola, Ciro Moretti, Simone Cigni e Rodolfo Lisi affrontano il tema del ricondizionamento muscolare nella CMT, nominando positivamente, per la prima volta, l’Attività […]
Un passo avanti nella ricerca sulla CMT2A: il potenziale dei biomarcatori e il valore del contributo dei pazienti
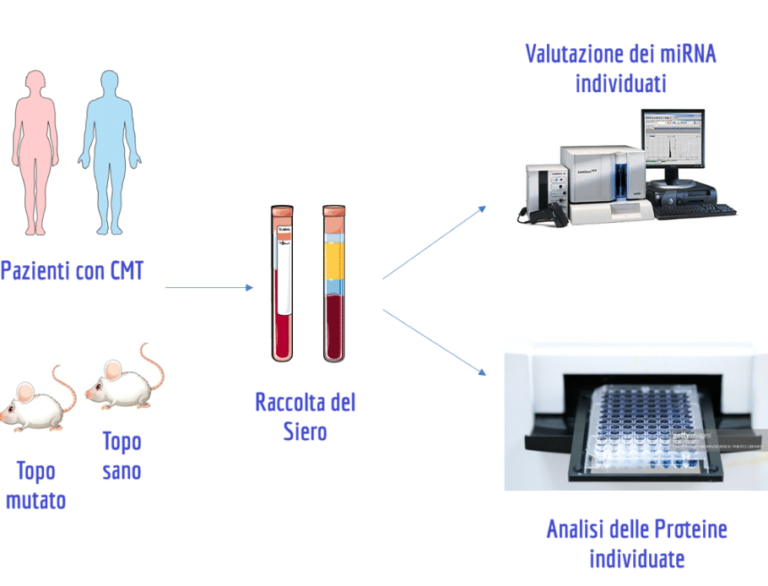
La ricerca scientifica sulla malattia di Charcot-Marie-Tooth di tipo 2A (CMT2A) ha raggiunto un traguardo promettente volto a migliorare la gestione clinica e lo sviluppo di nuove terapie. La patologia, essendo a progressione lenta, rende complesso monitorare l’efficacia dei potenziali trattamenti utilizzando solo le tradizionali valutazioni cliniche. Per questo motivo, un recente studio, pubblicato sulla […]
L’Intelligenza Artificiale al Servizio della Ricerca sulla CMT: Il Progetto Dancer
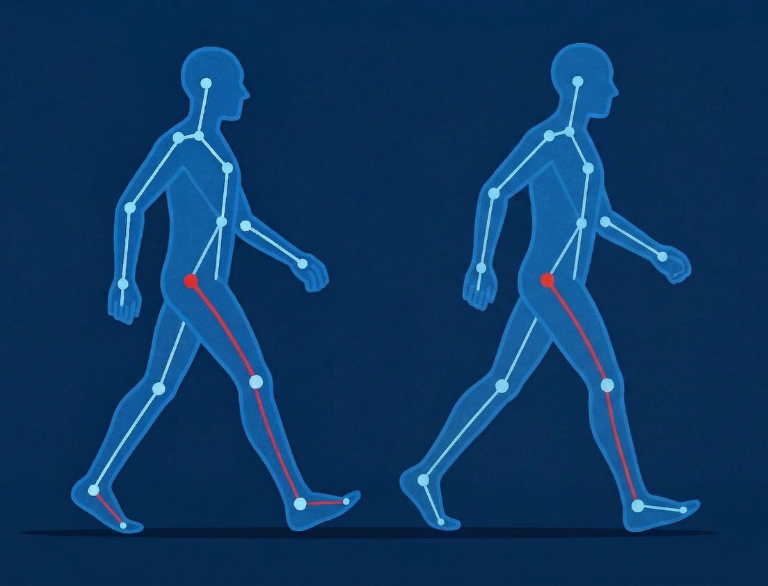
Convivere con la malattia di Charcot-Marie-Tooth (CMT) comporta sfide quotidiane, in particolare legate all’equilibrio e alla deambulazione. Storicamente, misurare l’evoluzione di queste difficoltà motorie nel tempo, o valutarne l’efficacia di un potenziale trattamento, ha rappresentato un ostacolo tecnico ed economico significativo per l’avvio e la gestione degli studi clinici. La Rivoluzione Digitale nei Trial Clinici […]