Charcot-Marie-Tooth: Aggiornamenti al 2026

Dalla diagnosi alle nuove terapie sperimentali: tutto quello che c’è da sapere sulla neuropatia ereditaria più comune.
La malattia di Charcot-Marie-Tooth (CMT) è una compagna di vita per molti di noi. Spesso ci identifichiamo con il pinguino, la nostra mascotte, per via di quell’andatura un po’ incerta e dondolante che ci accomuna. Ma oltre ai simboli, c’è una scienza che corre veloce.
Grazie a una recente revisione pubblicata su Nature Reviews Disease Primers (Gennaio 2026), possiamo offrirvi una panoramica aggiornata e rigorosa su cosa significa vivere con la CMT oggi e cosa ci riserva il domani.
🟢 IN BREVE: L’essenziale in un colpo d’occhio
Se hai poco tempo, ecco i punti fondamentali da conoscere:
- Diffusione: Colpisce almeno 17-20 persone ogni 100.000 nel mondo. La forma più comune è la CMT1A (duplicazione del gene PMP22).
- La Causa: È causata da mutazioni in oltre 130 geni diversi. Queste mutazioni danneggiano i “cavi elettrici” del nostro corpo (i nervi periferici), colpendo o il rivestimento isolante (mielina) o il filo conduttore interno (assone).
- I Sintomi: Si manifestano tipicamente con debolezza e atrofia muscolare che inizia tipicamente dagli arti inferiori e, nel tempo, interessa anche le mani (distribuzione “a calza e a guanto”).
- Gestione: Al momento non esiste una cura della malattia. Il trattamento è “di supporto”: riabilitazione, fisioterapia, uso di tutori e chirurgia ortopedica per migliorare la qualità della vita.
- Qualità della Vita: La CMT non riduce solitamente l’aspettativa di vita, ma incide sulla mobilità, sull’equilibrio e porta spesso fatica e dolore.
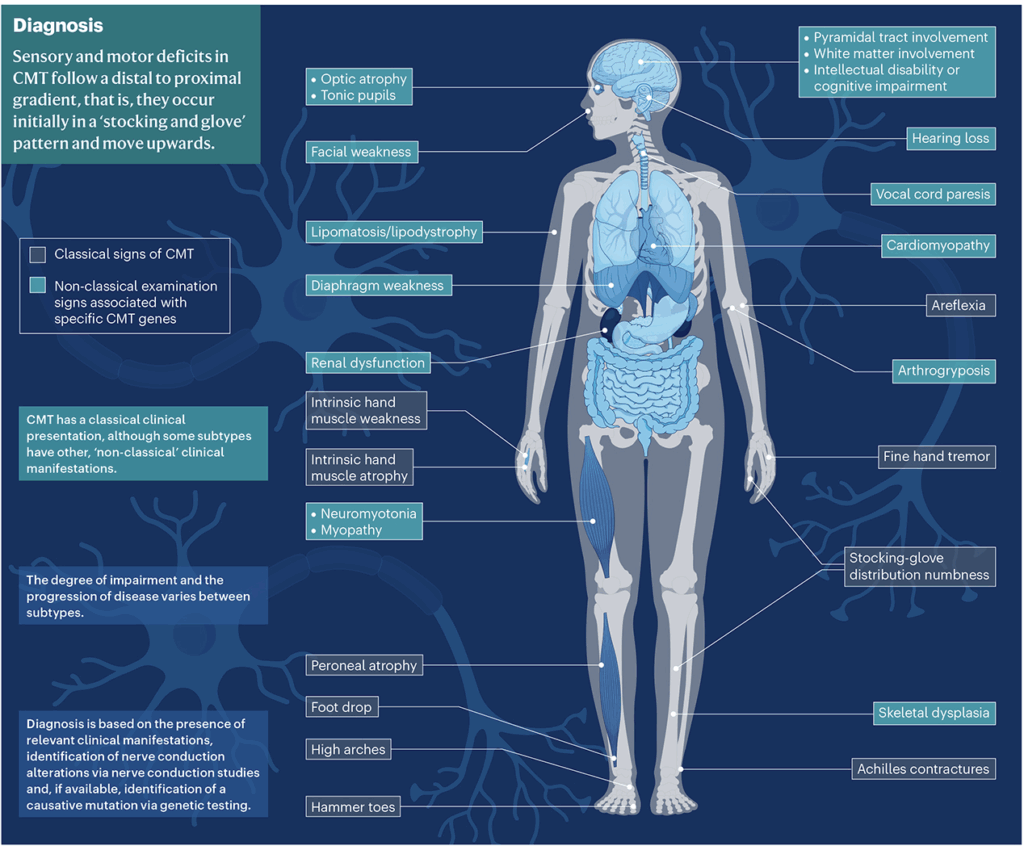
Mappa visiva della CMT: l’infografica mostra i sintomi classici (come il piede cadente e le dita a martello) e quelli “non classici” che possono apparire in forme più rare.
1. Che cos’è la CMT?
La Charcot-Marie-Tooth è un gruppo di neuropatie ereditarie. Immaginate i nervi come cavi elettrici che portano i comandi dal cervello ai muscoli e riportano le sensazioni (tatto, dolore) al cervello.
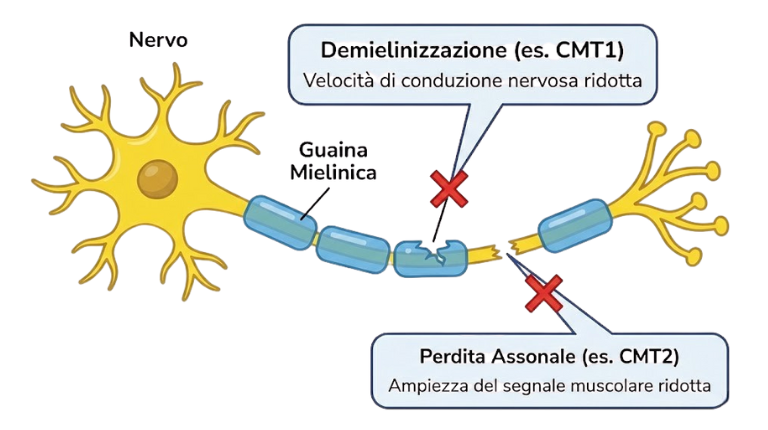
- Nella CMT1 (Demielinizzante), la guaina isolante, mielina, non funziona correttamente: il segnale viaggia troppo lentamente.
- Nella CMT2 (Assonale), si rovina il “filo di rame” interno: il segnale viaggia alla velocità giusta, ma è debole.
Il risultato è simile: i muscoli distali (mani e piedi) si indeboliscono progressivamente e la sensibilità diminuisce.
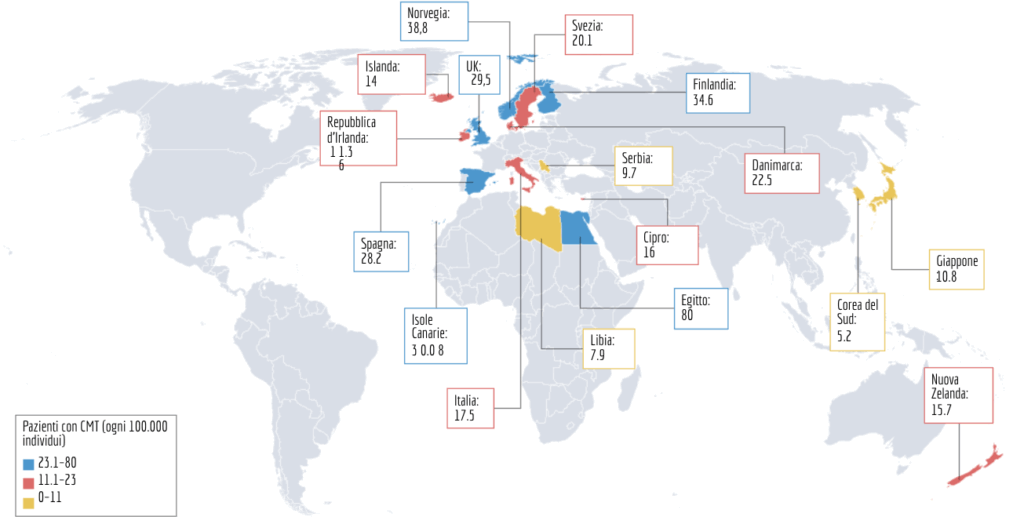
2. Quanto è diffusa? Focus sull’Italia
I nuovi dati del 2026 ci dicono che la malattia è molto più variabile di quanto pensassimo. Mentre la media globale è di circa 1 su 2.500 persone, ci sono differenze enormi tra i vari paesi:
- Nel Mondo: Si va dai picchi dell’Egitto (80 casi su 100.000) e della Norvegia (38,8), ai tassi più bassi della Corea del Sud (5,2).
- In Italia: La prevalenza stimata è di 17,5 casi ogni 100.000 abitanti.
- Attenzione: Questo numero è probabilmente una sottostima. Molte persone con sintomi lievi non ricevono mai una diagnosi.
- Il ruolo dei Registri: Il dato italiano è frutto del prezioso lavoro del registro Nazionale CMT.
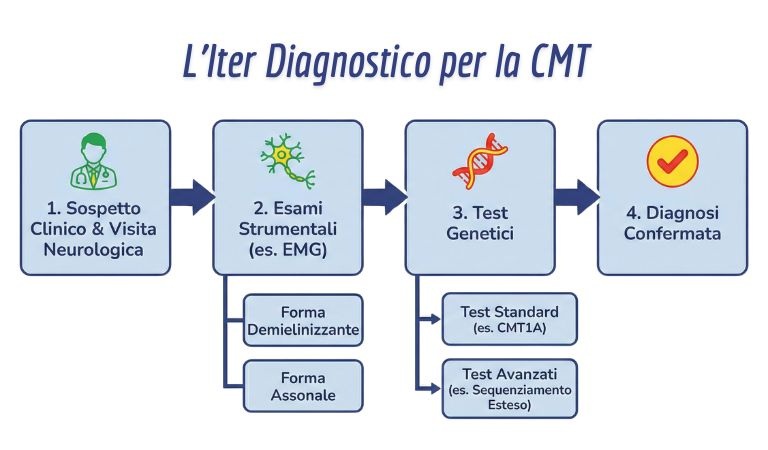
3. I Sintomi e la Diagnosi: Come riconoscerla
Oltre ai segni classici (piede cavo, dita a martello, gambe sottili “a cicogna”), la CMT può presentarsi in modi diversi a seconda del gene colpito.
La diagnosi oggi segue un percorso preciso:
- Visita Neurologica: Valutazione dei riflessi (spesso assenti) e della forza.
- Elettromiografia (EMG): Fondamentale per capire se è una forma demielinizzante (velocità lenta) o assonale.
- Test Genetici: Sono il cuore della diagnosi. Si cerca prima la mutazione più comune (CMT1A). Se negativa, si usano pannelli più ampi.
- Nuove Frontiere (Long-read Sequencing): Se i test standard non trovano nulla, dal 2026 si consigliano tecniche avanzate capaci di leggere “pezzi” di DNA lunghi, svelando mutazioni nascoste in geni complessi come SORD o RFC1.
4. Gestione Quotidiana: Cosa fare oggi
Non esiste ancora la pillola magica, ma “curarsi” significa prendersi cura del proprio corpo ogni giorno:
- Riabilitazione: L’esercizio fisico è sicuro e raccomandato. Rinforzare i muscoli (anche quelli prossimali come anche e tronco) aiuta a compensare la debolezza dei piedi.
- Tutori (Ortesi): Non vederli come una sconfitta, ma come strumenti di libertà. Aiutano a camminare meglio, inciampare meno e stancarsi meno.
- Chirurgia: Interventi mirati per correggere deformità ossee possono essere risolutivi per il dolore, ma affidatevi sempre a chirurghi esperti in malattie neuromuscolari.
- Gravidanza: È un tema importante. I sintomi possono peggiorare nel 16-38% delle donne durante la gravidanza, ma spesso tornano alla normalità dopo il parto.
5. Speranza dalla Ricerca: Le Terapie in Sperimentazione (Trial Clinici)
La review del 2026 elenca diverse terapie che hanno superato la fase di laboratorio e sono attualmente in fase di sperimentazione sull’uomo (trial clinici). Ecco le più promettenti, divise per meccanismo d’azione:
1. “Silenziare” il gene in eccesso (Per la CMT1A)
Nella CMT1A, il problema è avere “troppa” proteina PMP22. L’obiettivo è abbassare questo volume.
- EDK060 (ex DTx-1252): È una terapia basata su siRNA (piccole molecole di RNA) progettata per ridurre i livelli di PMP22. È attualmente in uno studio di Fase 1 per adulti con CMT1A.
- PMP22 “Silencers”: Altre strategie che usano oligonucleotidi antisenso (ASO) per “spegnere” l’RNA messaggero della PMP22 hanno mostrato risultati eccezionali nei modelli animali, invertendo i sintomi, e sono vicine ai test sull’uomo.
2. Ripulire le tossine metaboliche (Per CMT-SORD e HSN1)
Alcune forme di CMT sono causate dall’accumulo di sostanze tossiche nel nervo.
- AT-007 (Govorestat): Per la CMT-SORD. Chi ha questa forma accumula sorbitolo, che è tossico per i nervi. Questo farmaco inibisce l’enzima che produce sorbitolo. È in fase di sperimentazione avanzata.
- L-Serina: Per la HSN1. È un integratore orale che riduce i livelli di sfingolipidi tossici nel sangue. Un trial clinico è attualmente in corso (NCT06113055).
3. Migliorare la qualità delle proteine (Per CMT1A e CMT1B)
- IFB-088 (Sephin1): Nelle forme di CMT dove le proteine si ripiegano male (causando stress alla cellula), questa molecola aiuta le cellule a gestire lo stress (modulando la risposta UPR). Ha completato con successo la Fase 1.
4. Potenziare il muscolo (Per CMT1 e CMT2)
Invece di curare solo il nervo, si cerca di aiutare il muscolo a lavorare meglio.
- NMD670: È un inibitore di un canale del cloro specifico del muscolo scheletrico. L’obiettivo è migliorare la forza e la funzione muscolare sia nella CMT1 che nella CMT2. Un trial è appena terminato e a breve conosceremo i risultati (NCT06482437).
5. Terapia Genica e Cellulare
- Terapia Genica (AAV9): Per forme gravissime come la CMT2S (legata al gene IGHMBP2), si sta tentando di reintrodurre il gene sano tramite un virus vettore inattivato.
- Inibitori HDAC6: Questa classe di farmaci punta a migliorare il “trasporto merci” all’interno dell’assone, che è difettoso in molte forme di CMT2. Sperimentazioni sono in corso su adulti sani per valutarne la sicurezza.
Conclusioni
La strada verso la cura è tracciata e non siamo mai stati così vicini. Dalla diagnosi sempre più precisa alle terapie che correggono i difetti genetici, il panorama della CMT sta cambiando radicalmente.
Il consiglio per tutti noi “pinguini”? Restare attivi, mantenersi in forma e informati tramite canali affidabili come ACMT-Rete.
Fonte: Nature Reviews Disease Primers (Gennaio 2026) – “Charcot-Marie-Tooth disease and related neuropathies”.
Nota: Queste informazioni sono aggiornate al Gennaio 2026. È importante ricordare ai lettori che la partecipazione ai trial clinici deve essere sempre valutata con il proprio specialista di riferimento.