Congresso Società Nervo Periferico (PNS) 2023: Novità per la CMT

Scopri le novità relative alla malattia di Charcot-Marie-Tooth presentate al congresso della Società Nervo Periferico (PNS) 2023
Si è da poco conclusa a Copenaghen la Riunione annuale della PNS 2023: la Peripheral Nerve Society è la più grande società scientifica per le Neuropatie Periferiche al mondo. Durante l’incontro, sono state presentate le ultime ricerche internazionali nel campo delle neuropatie periferiche, con un focus group sulla malattia di Charcot-Marie-Tooth.
Abbiamo chiesto alla dott.ssa Valeria Prada, dell’Università di Genova, di creare un sunto di alcune delle ricerche più significative che riguardano direttamente la CMT.
Nuove terapie per la Charcot-Marie-Tooth
 Il dott. Davide Pareyson ha fatto il punto della situazione sulle numerose strategie di cura durante il focus group sulla malattia di Charcot-Marie-Tooth CMTR. Non esiste ancora un trattamento farmacologico efficace per la (CMT). L’attuale gestione si basa sulla terapia riabilitativa, sulla chirurgia per le deformità scheletriche e sul trattamento sintomatico. La sfida è trovare terapie che modifichino il decorso della malattia. Sono allo studio diversi approcci, tra cui il silenziamento genico (mediante ASO (nucleotidi antisenso), siRNA, CRISPR-Cas9 editing) per contrastare la sovraespressione del gene PMP22 nel tipo più frequente di CMT1A. PXT3003 è il composto nella fase più avanzata per CMT1A, poiché è in corso un secondo studio di fase III. La terapia genica per sostituire i geni mutati (in particolare nelle forme recessive associate a mutazioni con perdita di funzione) o inserirne di nuovi (ad esempio, il gene NT3) è in fase di sviluppo e sperimentazione su modelli animali. Nuovi approcci terapeutici mirano anche a sviluppare composti che agiscono su meccanismi importanti per diversi tipi di CMT. La modulazione della via della Neuregulina, proteina che determina lo spessore della mielina, è promettente sia per le neuropatie ipo-demielinizzanti che ipermielinizzanti; l’intervento sulla Unfolded Protein Response (risposta dovuta a proteine non correttamente ripiegate) sembra efficace per il recupero di proteine della mielina mal ripiegate come P0 nella CMT1B. Gli inibitori dell’HDAC6 hanno migliorato il trasporto assonale e i fenotipi in diversi modelli di CMT. Altre potenziali strategie terapeutiche includono il targeting di macrofagi, metabolismo lipidico e canale del sodio Nav1.8 nelle CMT demielinizzanti e il recettore P2X7, che regola l’afflusso di calcio nelle cellule di Schwann, nella CMT1A. Ulteriori approcci mirano a correggere le anomalie metaboliche, compreso l’accumulo di sorbitolo causato da mutazioni bialleliche nel gene della sorbitolo deidrogenasi (SORD) e di glicosfingolipidi neurotossici in HSN1.
Il dott. Davide Pareyson ha fatto il punto della situazione sulle numerose strategie di cura durante il focus group sulla malattia di Charcot-Marie-Tooth CMTR. Non esiste ancora un trattamento farmacologico efficace per la (CMT). L’attuale gestione si basa sulla terapia riabilitativa, sulla chirurgia per le deformità scheletriche e sul trattamento sintomatico. La sfida è trovare terapie che modifichino il decorso della malattia. Sono allo studio diversi approcci, tra cui il silenziamento genico (mediante ASO (nucleotidi antisenso), siRNA, CRISPR-Cas9 editing) per contrastare la sovraespressione del gene PMP22 nel tipo più frequente di CMT1A. PXT3003 è il composto nella fase più avanzata per CMT1A, poiché è in corso un secondo studio di fase III. La terapia genica per sostituire i geni mutati (in particolare nelle forme recessive associate a mutazioni con perdita di funzione) o inserirne di nuovi (ad esempio, il gene NT3) è in fase di sviluppo e sperimentazione su modelli animali. Nuovi approcci terapeutici mirano anche a sviluppare composti che agiscono su meccanismi importanti per diversi tipi di CMT. La modulazione della via della Neuregulina, proteina che determina lo spessore della mielina, è promettente sia per le neuropatie ipo-demielinizzanti che ipermielinizzanti; l’intervento sulla Unfolded Protein Response (risposta dovuta a proteine non correttamente ripiegate) sembra efficace per il recupero di proteine della mielina mal ripiegate come P0 nella CMT1B. Gli inibitori dell’HDAC6 hanno migliorato il trasporto assonale e i fenotipi in diversi modelli di CMT. Altre potenziali strategie terapeutiche includono il targeting di macrofagi, metabolismo lipidico e canale del sodio Nav1.8 nelle CMT demielinizzanti e il recettore P2X7, che regola l’afflusso di calcio nelle cellule di Schwann, nella CMT1A. Ulteriori approcci mirano a correggere le anomalie metaboliche, compreso l’accumulo di sorbitolo causato da mutazioni bialleliche nel gene della sorbitolo deidrogenasi (SORD) e di glicosfingolipidi neurotossici in HSN1.
CRISPR Cas9 e CMT
La dott.ssa Jae Young Lee ha presentato un nuovo approccio con la tecnologia CRISP CAS9 (editing genetico) in vivo su un modello murino di CMT1A, con l’intento di diminuire l’overespressione della proteina PMP22. Hanno testato questa tecnica su tre modelli animali (Topo C3, C22 e ratto), ma solo i topi C3 hanno mostrato dei miglioramenti. Pur condividendo diverse perplessità sull’utilizzo di questa tecnologia ancora immatura in vivo sull’uomo, si tratta del primo esperimento.
Una nuova classificazione per la CMT X-Linked (CMT1X)
Il dott. Chris Record dello UCL di Londra ha presentato una classificazione di GJB1, gene che codifica per la Connessina 32 e le cui mutazioni sono alla base della CMTX1, analizzando quasi 400 famiglie. Utilizzando diversi criteri, più stringenti, il loro modello ha permesso una classificazione più capillare (considerando frequenza allelica, fenotipo, segregazione e altre variabili genetiche) di queste forme, ipotizzando di proporre questa classificazione come standard.
Nuovi geni collegabili a forme di Charcot-Marie-Tooth
La dott.ssa Arnirola-Ricaurte ha presentato casi di 3 famiglie non correlati tra loro con mutazioni nel gene NDUFS6, che è un gene nucleare mitocondriale. Questo lavoro fornisce prove a sostegno del fatto che NDUFS6 sia un nuovo gene che causa la malattia per CMT, e che è necessario espandere lo spettro clinico dei disturbi mitocondriali correlati a NDUFS6. La dottoressa ha anche sottolineato l’importanza dello splicing, in quanto potrebbe differenziare tra un fenotipo letale e vitale.
La dott.ssa Sara Nagy ha invece parlato di varianti associate al gene RTN2 scoperte con exome e genome sequencing. In queste mutazioni è presente un sottotipo di pazienti che insieme alla spasticità presenta anche una neuropatia. Studi in vitro hanno evidenziato come sia presente un malfunzionamento nel reuptake del calcio (nel C. Elegans, utilizzato come modello sperimentale, un’inibizione del reuptake del calcio causa una regressione dei segni patologici), meccanismo sul quale si potrebbe intervenire per un trattamento di questa forma.

Una maggiore comprensione di MPZ
Il gruppo del Prof. Shy ha presentato un interessante lavoro sul funzionamento della proteina della Mielina 0 (MPZ), le cui alterazioni genetiche sono alla base della CMT1B e di alcune CMT assonali. Dalla letteratura si sa che la proteina può creare dimeri e tetrameri. I primi si verificano tra proteine di foglietti mielinici diversi, i secondi tra le proteine dello stesso “lato”. Le mutazioni di un particolare dominio possono interferire con la creazione dei dimeri e quindi potrebbero essere alla base di un nuovo possibile meccanismo di malattia.
Una via di trasporto importante
Il Prof. Schiavo del dipartimento di malattie neuromuscolari dell’UCL Queen Square Institute of Neurology, studia da tempo i passaggi chiave nel meccanismo di secrezione e assorbimento a livello delle sinapsi dei neuroni e il reclutamento dei complessi ligando-recettore nel trasporto assonale retrogrado. Questa via di trasporto essenziale, che fornisce una varietà di organelli e complessi molecolari al corpo centrale del neurone, è compromessa in diverse patologie del sistema nervoso come la malattia di Charcot Marie e la malattia del motoneurone.
Analisi del DNA con le nuove tecnologie: non banale
 La lectio magistralis della Prof.ssa Kennerson, ricercatrice genetista australiana, ha mostrato quanto incredibilmente complessa sia la ricerca delle mutazioni che conducono allo svolgersi della malattia. Infatti, se ad oggi abbiamo tecniche sempre più avanzate che “leggono” il genoma umano velocemente, tuttavia ci sono ancora molte cose che non sappiamo del genoma, come le parti che non codificano per proteine ma che, se mutate, interferiscono con la parte non codificante e quindi generano la malattia (e per questo ancora più difficili da trovare). Con la nuova generazione di sequenziatori possiamo trovare mutazioni nelle regioni codificanti e non. La faccenda si complica ulteriormente perché se fino ad oggi si considerava il codice genetico come un qualcosa in “da leggere” come una pagina, diventa necessario prendere coscienza che il DNA è ancora più complesso e ha una struttura tridimensionale che può subire delle mutazioni (interfusion transcript). In conclusione, sicuramente la parte della genetica tradizionale è la nostra comfort zone che deve essere sempre condotta nella pratica clinica, ma se non si riesce a trovare la causa di una neuropatia, si dovrà cercare più a fondo, in un universo che è ancora misterioso e poco conosciuto.
La lectio magistralis della Prof.ssa Kennerson, ricercatrice genetista australiana, ha mostrato quanto incredibilmente complessa sia la ricerca delle mutazioni che conducono allo svolgersi della malattia. Infatti, se ad oggi abbiamo tecniche sempre più avanzate che “leggono” il genoma umano velocemente, tuttavia ci sono ancora molte cose che non sappiamo del genoma, come le parti che non codificano per proteine ma che, se mutate, interferiscono con la parte non codificante e quindi generano la malattia (e per questo ancora più difficili da trovare). Con la nuova generazione di sequenziatori possiamo trovare mutazioni nelle regioni codificanti e non. La faccenda si complica ulteriormente perché se fino ad oggi si considerava il codice genetico come un qualcosa in “da leggere” come una pagina, diventa necessario prendere coscienza che il DNA è ancora più complesso e ha una struttura tridimensionale che può subire delle mutazioni (interfusion transcript). In conclusione, sicuramente la parte della genetica tradizionale è la nostra comfort zone che deve essere sempre condotta nella pratica clinica, ma se non si riesce a trovare la causa di una neuropatia, si dovrà cercare più a fondo, in un universo che è ancora misterioso e poco conosciuto.
La composizione della Mielina nella Charcot-Marie-Tooth
Il dott. Davide Visigalli dell’Università di Genova ha presentato un lavoro sullo studio della composizione lipidica nella mielina dei ratti con CMT1A, dimostrando che questa è molto diversa rispetto ai ratti “sani”. Inoltre, la maturazione normale della mielina dipende da un costante cambiamento di questo assetto lipidico. Nei ratti con CMT1A questo cambiamento di assetto lipidico sembra non esserci, e producono una mielina più immatura e meno capace di isolare l’assone nel processo di conduzione dell’impulso nervoso.
La Risonanza Magnetica per valutare la gravità della CMT e l’efficacia dei trattamenti
Un lavoro interessante è stato portato dal dott. Luke O’Donnel del Queen Square di Londra riguardante la possibilità di utilizzare la RM come biomarker nei nuovi futuri trial clinici e portando un esempio di protocollo da utilizzare. Lo studio è ancora in corso (ed è stato rallentato nel reclutamento dal Covid-19), ma ha dimostrato una buona possibilità di standardizzazione della procedura in diversi centri internazionali e sembra che la RM dell’arto inferiore possa dare risultati interessanti e relativamente significativi se confrontati con altre scale di valutazione (CMTES) o biomarcatori (NFL).
Un nuovo approccio via siRNA testato su topolini
La DTx Pharma ha presentato un nuovo metodo di trattamento della CMT1A, tramite un siRNA (small interfering RNA) coniugato a un acido grasso, una molecola che entra nelle cellule di Schwann e silenzia il gene che produce PMP22. Hanno testato questa molecola sul topo C3 transgenico per duplicazione di PMP22 e hanno visto che i nervi sembrano rimielinizzare e si ha un aumento delle velocità di condizione motorie oltre che della forza dopo 12 mesi. Il farmaco è stato dato una volta al mese per tre mesi. Non è ancora però chiaro quanto prolungare la terapia, eventuali effetti collaterali, seguiremo con attenzione il futuro di questo potenziale candidato farmaco.
Lo studio INSPIRE per le CMT-SORD
Il Prof. Shy ha presentato lo studio INSPIRE, che riguarda le forme di CMT-SORD. Lo studio coinvolge diversi centri a livello mondiale in un trial di fase 3 multicentrico randomizzato controllato in doppio cieco. Lo studio è stato disegnato per verificare l’efficacia di un farmaco, AT-007 e per testare la somministrazione a lungo termine tramite diversi outcome clinici. Lo studio è ancora in corso, attendiamo con le dita incrociate i risultati.
Il ruolo della giunzione neuromuscolare
ESTABLISH (NCT04980807), uno studio osservazionale lanciato da NMD Pharma nel 2021 per studiare come la CMT si manifesti a livello di giunzione neuromuscolare (NMJ), il punto di contatto tra nervi e muscoli, ha dimostrato che la trasmissione dei segnali tra i nervi e i muscoli è compromessa nelle persone con Charcot-Marie-Tooth di tipo 1 o 2 e il grado di compromissione sembra essere correlato alla gravità della malattia. Lo studio ha fornito informazioni importanti su come misurare in modo affidabile la trasmissione a livello di giunzione neuromuscolare, misura che potrebbe essere usata in future sperimentazioni cliniche e per sviluppare nuovi trattamenti.
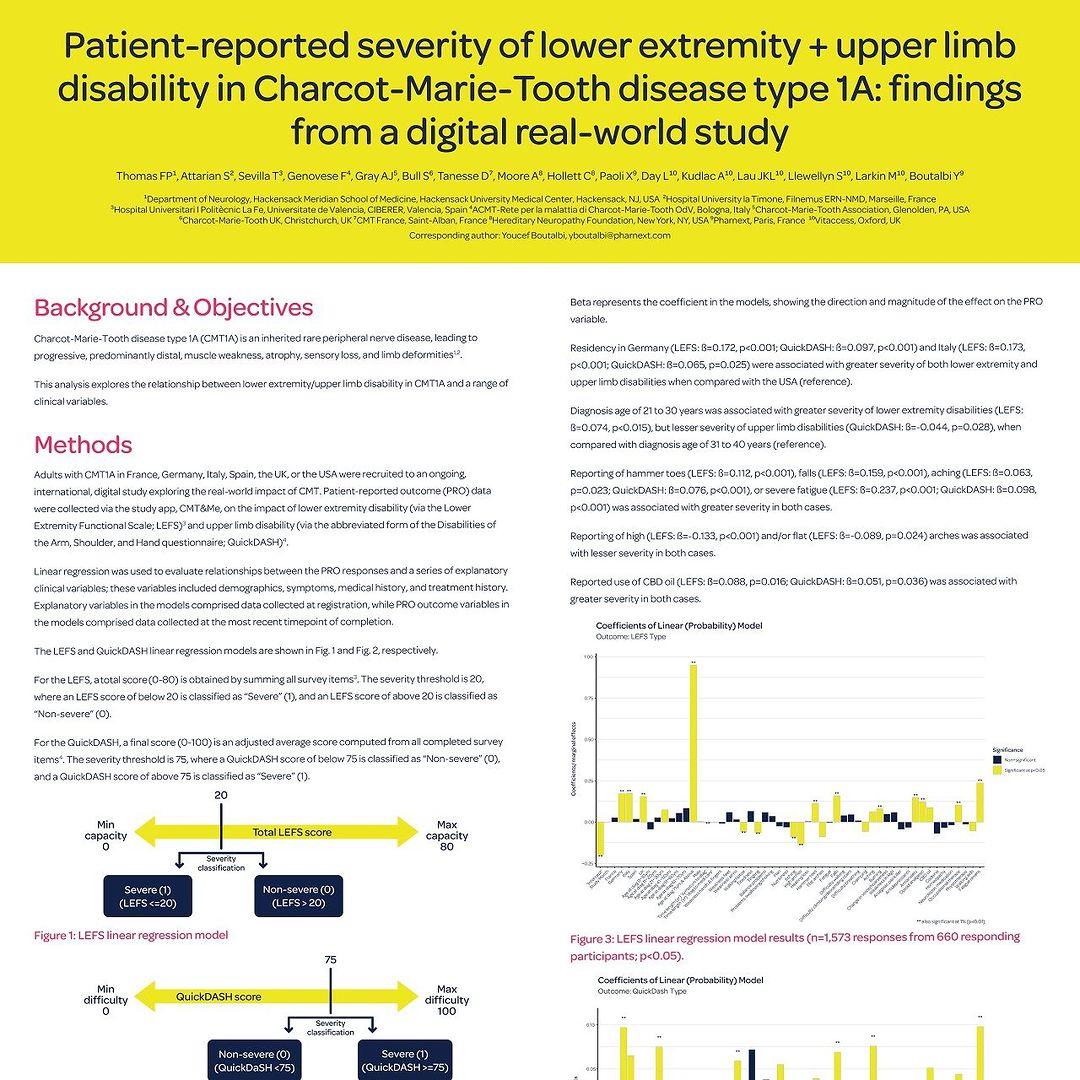
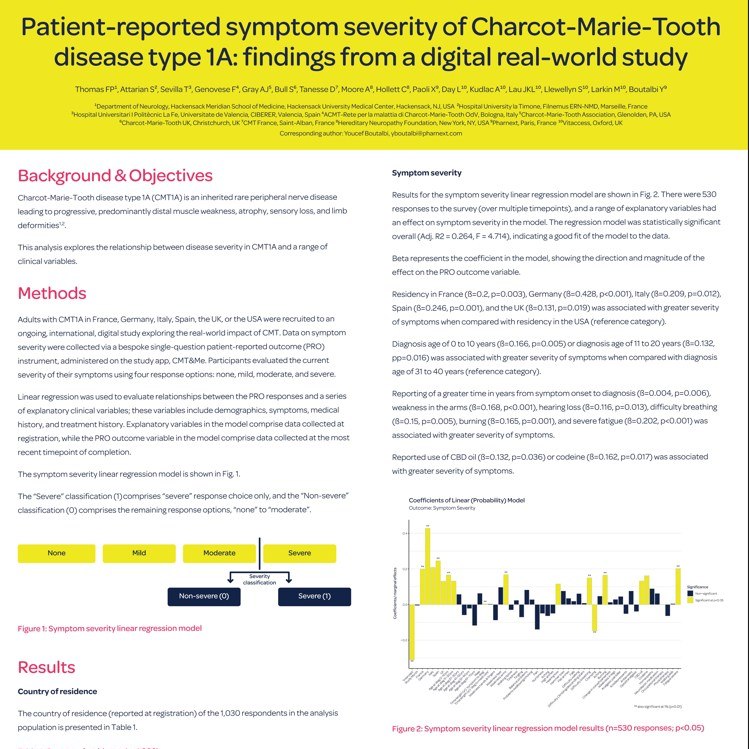
Due Poster dello studio CMT&Me
Il team dello studio internazionale CMT&Me ha presentato due interessanti poster che aiuteranno a comprendere quanto la Charcot-Marie-Tooth influenzi la vita delle persone, realtivamente ai sintomi e difficoltà alle mani e il loro impatto sul paziente:
Premiazione alla carriera per il Prof. Vincent Timmerman
Durante l’evento, la società per il nervo periferico ha premiato il Prof. Vincent Timmerman che per primo, insieme al Prof. De Jonghe, ha descritto la duplicazione del gene per PMP22 come causa genetica scatenante della CMT1A, di fatto permettendo la comprensione delle cause molecolari e la diagnosi di questa forma di Charcot-Marie-Tooth.

Da sinistra il dott. Shy, la Prof. Reilly, il Prof. Timmerman e il dott. Zuchner

Università degli Studi di Genova